中国首款新型冠状疫苗进入临床试验,比预期提前近一个月
中国进度最快的新型冠状病毒疫苗已进入临床试验志愿者招募阶段。今天(3月17日)上午,康希诺生物-B(06185.HK)发布公告称,公司与军科院生物工程研究所联合开发重组新型冠状病毒疫苗(腺病毒载体),目前已递交新药临床试验预审评申请,并启动健康志愿者预招募。
官方消息验证了康希诺的进展。今天下午,国务院联防联控机制发布会上,未点名地介绍了中国疫苗研发的最新进展——“第一批确定的9项疫苗研发任务,目前都已完成大部分临床前研究工作,包括动物有效性和安全性试验,预计大部分研发团队在4月份都能完成临床前的准备工作,并陆续启动临床申请、临床试验”。
发布会上同时披露:“我国已经有研发进展比较快的单位向国家药监局滚动提交临床试验申请材料,并且开展临床试验方案的论证、招募志愿者等相关工作,待国家药监局按有关法律法规审批以后开展临床试验。我国疫苗研发进展属于国际先进行列,不会慢于国外”。
这一进展比预期进入临床阶段的时间提前了近一个月。 在3月6日于湖北武汉举行的国家新闻办公室新闻发布会上,中央指导小组成员,国务院副秘书长丁向阳给出了最早的数据,预计疫苗的最新研究在4月中或更早的时间将报告用于临床试验。
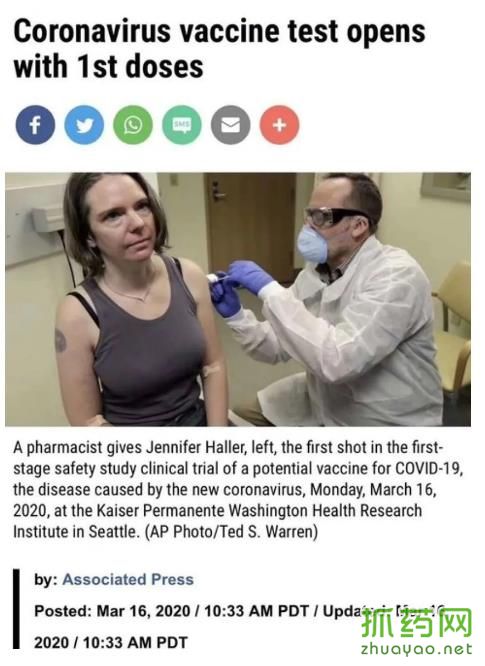
从全球来看,进展最快的是美国Moderna公司,3月5日该公司疫苗“mRNA-1273”临床试验获批,昨天(3月16日),4名受试者在西雅图接受了疫苗注射。
康希诺和Moderna疫苗原理不同,但都是最新的技术
康希诺的联合开发方正是此前因一张配文为“疫苗第一针,院士先试”的图片而备受关注的陈薇院士团队,尽管后来的报道证实这张照片中注射的并非新冠疫苗,但当时确实已有进展。当晚《新闻联播》指出“由军事医学研究院陈薇院士领衔的科研团队在新冠肺炎疫苗研制方面取得了重要阶段性成果”,不过并未指明具体的成果。
直到今天,康希诺公告发布,该公司和军科院联合开发的疫苗Ad5-nCoV采用基因工程方法构建,以复制缺陷型人5型腺病毒为载体,可表达新型冠状病毒S抗原,用于预防新型冠状病毒感染引起的疾病。
这不是军科院第一次与康希诺合作。此前双方合作用同样的技术成功研发了埃博拉疫苗,并于2017年在国内完成审批上市流程。
一位熟悉陈薇团队的传染病学家告诉记者,此次研发新冠疫苗,继续沿用了埃博拉疫苗式的复制缺陷型人5型腺病毒作为载体。“腺病毒载体就像是酒瓶,我们现在把里面的白酒(埃博拉病毒)换成红酒(SARS-CoV-2病毒)了,而外面的酒瓶起到的保护作用是一样的。”
具体来讲,重组病毒载体疫苗是指一种活载体疫苗技术,也是最新一代的疫苗技术。其作用机制是,以病毒作为载体,将保护性抗原基因重组到腺病毒基因组中,使用能表达保护性抗原基因的重组腺病毒制成的疫苗。以埃博拉疫苗为例,在病毒载体(腺病毒)中加入表达可供埃博拉病毒进入细胞的埃博拉糖蛋白EBO-GP的基因,病毒在细胞内表达出了减毒的埃博拉病毒,能够刺激B细胞及T细胞以诱发免疫力。
感染细胞时,因为不整合到染色体中,激活致癌基因或插入突变等风险低,生物安全性高。根据这位传染病学家的估计,从现在开始算,到拿到上市许可证,哪怕在应急状态下,也至少还需要一年时间。
从疫苗原理和工艺上区分,疫苗主要可以分为传统的灭活、减毒疫苗;亚单位疫苗/ 基因重组蛋白疫苗;病毒载体疫苗以及核酸疫苗(mRNA疫苗和DNA疫苗)。
康希诺、军科院的重组病毒载体疫苗和Moderna的核酸疫苗,都是最新的技术。
核酸疫苗技术目前在全球范围内还没有成功上市的疫苗先例,与病毒载体疫苗技术用了病毒作为载体不同,mRNA技术将编码S蛋白的基因直接注入人体,利用人体细胞产生S蛋白,刺激人体产生抗体。
目前来看,Moderna公司的疫苗是进展最快的。3月5日, Moderna宣布,美国食品药品监督管理局(FDA)已完成对该公司研制的新型冠状病毒mRNA疫苗——“mRNA-1273”的审查,批准其进入临床试验。
据美联社报道,3月16日,4名受试者在华盛顿州西雅图市“凯撒医疗集团华盛顿健康研究中心”(Kaser Permannte Washinton Helth Reserch Institute)接受了疫苗注射。43岁的Jennfer Haller是第一个接种疫苗的志愿者,在mRNA-1273疫苗的临床1期试验中,共有45名志愿者,他们将在一个月内被注射两次剂量。
疫苗试验如何展开?以埃博拉疫苗为例
同药品研发过程一样,进入临床试验阶段之后,疫苗也需要经历三期临床试验,作为预防性疫苗,三期试验均以健康人群为受试对象。
临床1期通常需要20-80位试验对象,以评估疫苗的安全性,确定疫苗引起的免疫反应类型和程度。临床2期试验研究候选疫苗的安全性、免疫原性、建议的剂量、免疫接种计划和接种方法,由几百人组成的受试者小组参与。本阶段的目的是更详细地评估与1期相比的剂量和给药计划。在临床2期取得成功的基础上,候选疫苗将继续进入3期试验,试验人群从3千人至数万人不等。此阶段关键目标是评估疫苗的安全性和有效性。
而在紧急情况下,会加快审批流程,简化试验对象数量。根据一家疫苗公司负责人的估计,新冠疫苗只要在临床3期阶段能看到30-50例有效,就可以进入上市流程了。
具体地,临床阶段试验如何展开,我们可以回顾一下该团队此前的埃博拉疫苗试验过程,以作参考。
在康希诺的招股书中,详细记载了埃博拉疫苗的临床试验过程:
第一阶段为国内临床试验(Ia期临床试验),针对的是中国受试人群。研发团队与江苏省疾控中心合作,在泰州完成了对120名年龄在18至60岁的健康成年人的随机双盲、剂量递增、安慰剂对照临床试验。60名受试者按照2:1的比例被随机分配接种低剂量或安慰剂对照。在最初低剂量和安慰剂对照组中显示安全性和耐受性七天之后,另外60名受试者按照2:1的比例被随机分配接种高剂量或安慰剂对照。
试验结果证实了疫苗的安全性和免疫原性,接种后14天达到预期免疫效果,28天抗体水平达到峰值。
第二阶段依旧为国内临床试验(Ib期临床试验),针对的则是在华非洲人群。研发团队与浙江大学第一附属医院合作,受试者为61位年龄在18-60岁的健康非洲人。研究同样表明了疫苗的安全性和免疫原性。
比较泰州临床和杭州临床的检测结果,并未发现非洲及亚洲种族群体之间在免疫反应上存在种族或人种差异。在得出这一结果后,研发团队带着疫苗到非洲去。
于是临床试验进入了第三阶段(II期临床试验),也就是在非洲展开了境外试验,针对的是非洲人群,在塞拉利昂对500名年龄为18至50岁的健康成人进行的单中心、随机、双盲及安慰剂对照的试验。参与者按2:1:1的比例依次入组并随机分配接种高剂量疫苗、低剂量疫苗或安慰剂。
试验结果表明,自接种后第14天起,高剂量组及低剂量组均至少有96%的受试者中检测到抗体应答,并于第28天达至高峰。对健康的塞拉利昂成人而言,Ad5-EBOV具备安全性及令人满意的免疫原性。然而,抗体应答的持续时间较短,增加了初免-加强免疫接种的需要。
至2017年,由于处于埃博拉流行间歇期,难以提供大样本量人群有效性数据,Ⅲ期临床试验无法开展。针对这一状况,药审中心加快审评进程,根据当时已有研究得到的安全性和免疫原性数据,批准了重组埃博拉病毒病疫苗的有条件上市。上市后,重组埃博拉病毒病疫苗仅作为国家储备,用于疫情发生时的应急处理,并开展Ⅲ期临床试验。
疫苗直接关系世界疫情的下一步走向
至于疫苗最终能否顺利问世,“一方面与疫苗本身能否顺利研发成功相关,包括能否通过安全性的挑战;是否有很好的应答率和免疫原性,确实能在人体内产生免疫抗体;对疫苗目标人群是否有足够的保护效果;以及疫苗本身是否有持久性”,北京大学公共卫生学院卫生检验学系研究员、系主任崔富强分析称。
另一方面,也与疫情的发展相关。新冠病毒的发展有两种可能,一是像SARS一样,从此无踪无影,如果在临床试验阶段,疫情已经结束,病毒已经消失,那就无法继续开展临床试验,因为打了疫苗的实验组和没有打疫苗的对照组都没有机会感染病毒,就无法证明疫苗的有效性,试验也就无法科学开展。这种情况下做疫苗的可行性就不存在,理论上来说就不应该去做。二是可能这种病毒将来会重复出现,再次在人群中流行,那么做疫苗的必要性就非常大了。
至于病毒会否在将来重复出现,还有待时间的检验。“比如说这次是在11月以后到现在流行,如果说我们把它时间段看成每年的11月到3月之间流行的这么一个大趋势的话,那么它应该在每年这个时候出现。需要几年的数据才能说话,现在我们下结论,还为时过早。”崔富强表示。
而在今天国务院联防联控机制发布会上,教育部科技司司长雷朝滋说:疫苗作为疫情防控最有效的医学手段,可以有效的阻断病毒传播,不仅对中国取得抗击新冠疫情的最终胜利、稳定经济形势、有序复工复产具有重要的作用,而且直接关系世界疫情的下一步走向。
